脊髓性肌萎缩症( spinal muscular atrophy,SMA)是婴幼儿时期最常见的致死性神经遗传性疾病。该病为常染色体隐性遗传,主要致病基因位于5q 11-13上的SMN(survival motor neuron) 基因。该致病基因的人群携带率1/50~ 1/42, SMA的人群发病率为10000~1/6000。SMA病变主要累及脊髓前角α运动神经元,临床上主要以肌无力、肌张力下降、肌萎缩、腱反射减弱或消失为主要表现,通常近端重于远端,下肢重于上肢,疾病呈对称性进展,最终丧失运动能力,可导致死亡。
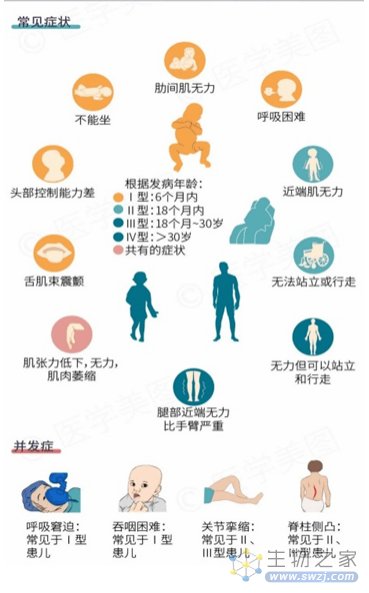
临床分型
自 SMA 命名以来,该病的诊断与分型一直以临床表现为主。 近年来,基因检测成为该病的一线诊断方法。 不同类型间临床表现不一致,严重程度也差异显著,其中1型SMA患儿最为严重,多在2年内死亡,2、3型患儿病死率低但容易并发各种畸形。
生物学相关标志物
1.NAIP 基因的缺失被证实与 SMA 严重程度呈负相关。 尤其当 SMN1⁃SMN2⁃NAIP 三者联合缺失时,SMA 发病年龄更早、病死率更高。
2.血浆和脑脊液的磷酸化神经丝重链(pNF⁃H)浓度与疾病严重程度呈正相关。
3.血肌酐水平可反映 SMA 严重程度,病情越重,血肌酐水平越低。
4.动物实验表明,SMA 小鼠存在睾丸发育不良、生殖功能减退表现,少许男性 SMA 患者也有睾丸功能低下的报道,但性激素水平监测作为该疾病的生物标志有待进一步研究。
治疗
SMA患者除肌无力外,多有呼吸系统受累,SMA 1 型、2 型的病例早期即可有心脏、肝脏、骨骼等受累,男性可有泌尿生殖系统受累,部分还表现有凝血功能异常。因此SMA综合管理共识主要包括营养支持、呼吸支持、康复训练、矫正继发性的骨关节畸形、预防感染等。
(1)基因修饰与基因治疗
Nusinersen是一种反义寡核苷酸,通过阻断 SMN2基因内含子7剪接沉默子 N1序列(ISS⁃N1)与 hnRNPA1/A2结合的方式保留第7外显子,提高全长 SMN 蛋白的表达。 通过Nusinersen鞘内注射治疗 可对早期1型,晚发2 型、3 型SMA患者均有不同程度的改善。 在自2016年以来,该药先后被美国、欧洲、加拿大批准用于脊髓性肌萎缩症的治疗,并于2019年在中国正式上市。
Zolgensma是 SMA 的首个基因治, 主要通过AAV(腺病毒) 介导引入外源性SMN基因,直接增加体内SMN蛋白的表达。 Ⅱ期临床试验显示,经过治疗的1型 SMA 患者运动评分显著提高。治疗后的 SMA 患者在肺炎的患病率、营养支持及住院需求方面也明显下降。2019 年 5 月经过美国 FDA 批准用于 1 型 SMA的治疗。
Risdiplam(RG7916、RO7034067)是一种口服给药的小分子 SMN2 前 mRNA 剪接修饰剂,可以增加 SMN2 在中枢神经系统和外周器官中分布并增加 SMN 蛋白浓度。该药已完成I期临床试验,25 名 18~45 岁间健康成年男性参与试验,最终结果证实 Risdiplam 受食物及肝酶影响小,组织分布好,安全性及耐受性好。
Exspe U1snRNAs 是一种特异性作用于外显子的小分子 RNA,通过 AAV9 病毒介导与 SMA 缺失的第 7 外显子下游的内含子序列相结合,使7号外显子得以保留,提高 SMN 蛋白水平,改善 SMA 症状,可作为潜在的药物治疗靶点。 目前该药研发尚停留在动物实验阶段。
(2) 其他药物
沙丁胺醇也可通过激活 B, 肾上腺素能受体-PKA 途径抑制泛素介导的 SMN 降解,增加全长蛋白表达,持续性口服沙丁胺醇治疗可提高患儿外周血中 SMN2 全长蛋白水平,改善其运动能力,其中呼吸肌的功能和力量亦可明显增强。
Olesoxime(TR019622)是胆固醇类化合物,通过减少凋亡因子的释放而实现神经保护和神经再生。目前Ⅱ期临床试验通过对 165 例 SMA 患者 2 型 3 型进行为期 24 月的治疗随访,发现 Olesoxime 治疗组在 MFM 量表 D1+D2 评分高于对照组(P<0.05),同时其安全性及耐受性再次得以证实。
左乙拉西坦是一种抗癫痫药,可通过抑制神经细胞凋亡,恢复细胞线粒体功能,减轻 SMA 患儿的临床症状,提示可作为 SMA 辅助治疗的候选用药,但该研究尚处于细胞研究阶段。
治疗评估
对于 1 型 SMA 患儿,常用生存时间、CHOP-INTEND 量表、HINE2 、HFMSE 量表评估其治疗效果。CHOP-INTEND 运动评估量表,主要包括 16 个项目(0~64 分),可以反映患儿疾病严重程度。HINE2 主要用于评估患儿的发育,适用于 2~24 月龄儿,包括抬头、独坐、抓握,蹬踢、翻身、爬行、站立、行走8个项目。而 HFMSE 运动功能评价量表(MFM)评分常被用于 2型 3型 SMA ,包括 33 个项目,每个项目 0~2分,总分 66 分,常作为临床试验的衡量标准之一。
结语
脊髓性肌萎缩症作为一种罕见的、高致死性的单基因遗传病,部分发达国家将其纳入新生儿筛查,但由于我国人口基数大、经济发展水平欠均衡、筛查成本较高,暂未纳入新生儿筛查。有 SMA 家族史的家庭,建议完善产前基因诊断,以提高优生优育。
近两年70万元/针的诺西那生钠注射液降低至3.3万并纳入医保给无数患儿家庭带来福音,但其背后的经济负担依然严重,多学科综合管理仍是我国 SMA 患者的主要治疗方案。 因此,早期生物标志物的出现是否会导致新的发病模式的发现、是否会引起临床分型的调整,仍有待进一步的观察随访,基因修饰及基因治疗药物的安全性及有效性也有待进一步证实。
参考文献
[1] 杨贇滢,洪思琦.儿童型脊髓性肌萎缩症诊治研究进展[J].儿科药学杂志,2022,28(12):55-58.DOI:10.13407/j.cnki.jpp.1672-108X.2022.12.015.
[2] 窦攀,熊晖,李融融等.脊髓性肌萎缩症患者的营养管理[J].中国实用儿科杂志,2022,37(10):748-754.DOI:10.19538/j.ek2022100608.