DNA是一种相对稳定的有机分子,但仍然会受到来自各种外源性压力(例如紫外线照射、电离辐射和化学暴露)以及内源性因素(例如复制错误、细胞代谢和氧化应激)的不断攻击,最终会导致单链DNA断裂(SSB)或双链DNA断裂(DSB)。
因此,细胞进化出了一个复杂的生化途径系统来处理这种威胁,统称为DNA损伤应答(DNA-damage response,简称DDR),以防止有害的突变被继续传递。
“合成致死”的概念用于描述两个功能性基因同时失活导致细胞死亡的现象,随着PARP抑制剂的成功,“合成致死”领域引起大家广泛的关注。在伴有DDR某条通路缺陷的肿瘤细胞中,DNA修复将高度依赖于其他旁路途径,而若此时这些通路被药物再次抑制则会产生“合成致死”效应。利用这一效应,DDR成为抗肿瘤药物的研发靶点。

DDR通路
DDR是生物的基本生理机制之一,这一机制旨在保护生物的基因组。2015年9月8日,DNA损伤应答发现者因此获得2015年拉斯克奖基础医学研究奖。
DNA损伤应答机制重要而复杂,主要通过两种手段,即DNA损伤修复和细胞凋亡,来保护其正常的生理功能和稳定的遗传性状(图1)。
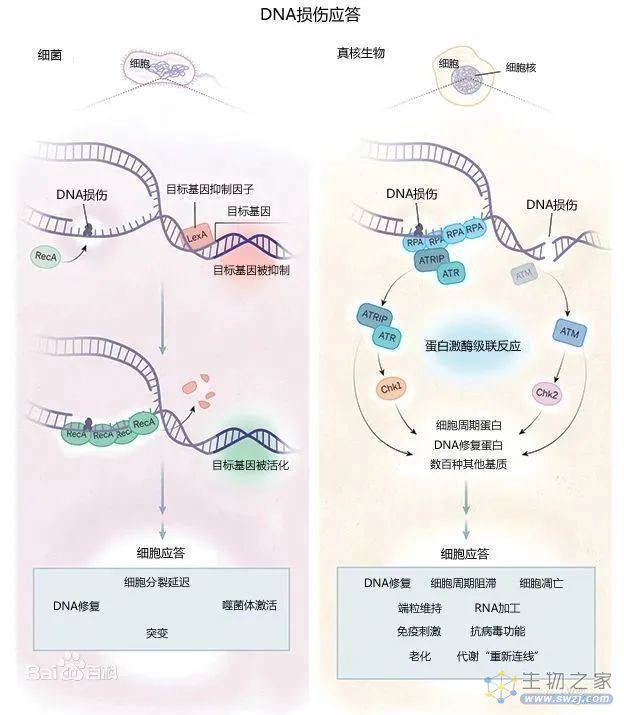
图1. DNA损伤应答,图源:百度百科
DNA损伤修复有非同源末端连接(NHEJ),同源重组(HR),错配修复(MMR),核苷酸切除(NER)等,识别DNA损伤和尝试修复DNA[1]。如果修复成功,细胞恢复复制;否则,就会触发细胞程序性死亡或衰老机制。
双链DNA断裂(DSB)可以通过两种主要机制进行修复:BRCA1/2介导的同源重组(HR)和典型 DNA-PKcs介导的非同源末端连接(c-NHEJ)。
HR被认为是一种准确的DSB修复途径,因为它依赖于细胞周期S/G2期细胞的姐妹染色单体作为DNA合成和DSB修复的模板。因此,HR能够修复DSB,主要在增殖细胞中。
DDR过程中涉及很多激酶,如RAD51,ATM,ATR,CHK1,WEE1,PKMYT1和DNA-PK等,它们的异常与很多疾病相关,它们也是合成致死领域研究的热门靶点。
临床靶向DDR激酶治疗有ATM抑制剂,DNA-PK抑制剂,ATR抑制剂,CHK1抑制剂,WEE1和PKMYT1抑制剂等,除此之外,还有一些新兴的疗法和新的靶点选择,如POLQ抑制剂针对HR缺乏的肿瘤,USP1抑制剂针对BRCA1/2缺失的癌症,RAD51抑制剂和靶向于微卫星不稳定性癌症中的解旋酶WRN等。本文主要介绍一下DDR通路相关激酶ATR,ATM,DNA-PK,CHK1,WEE1和POLQ抑制剂。
ATR和ATM抑制剂
当发生细胞DNA损伤时,ATM和/或ATR可以激活DDR修复途径,以确保细胞活力或细胞凋亡。ATR在被RPA(复制蛋白A)包被的单链(ss)DNA激活后,ATR的监管伙伴,即ATR相互作用蛋白(ATRIP),将直接与RPA结合,导致ATR定位到DNA损伤部位。最后,ATR-CHK1信令级联导致G2-M期的细胞周期停滞,为修复DNA损伤提供时间。ATM通过MRN复合物(MRE11-RAD50-NBS1)响应双链DNA断裂(DSBs)(图2)[2]。
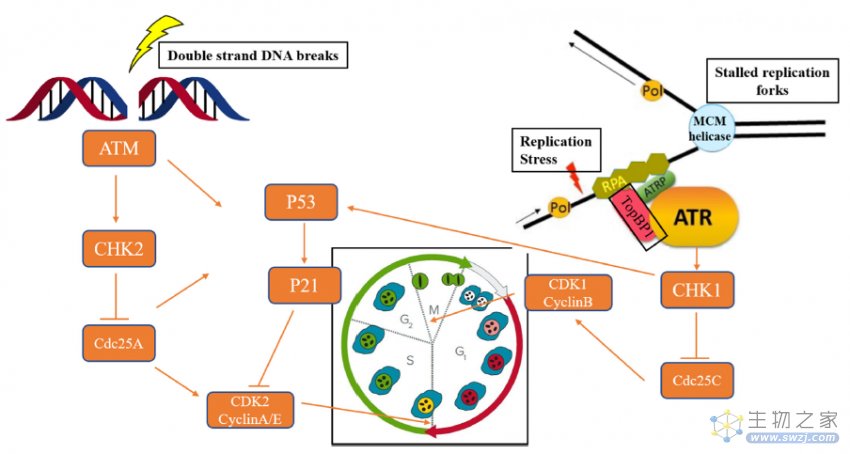
图2. ATM和ATR在DDR信号通路中的生物学作用
ATR相关抑制剂目前进入临床的有Berzosertib,Ceralasertib,M4344,M1774,ATRN-119,RP-3500和ART-0380。
Berzosertib(VE-822, M6620, VX-970)是第一个进入临床试验的ATR 激酶抑制剂, 由福泰制药(Vertex Pharmaceuticals)开发(图3)。它对ATR 激酶有很强的抑制活性, 相对于DNA-PK,Berzosertib 对ATR 的选择性比对DNA-PK的选择性超过4,000倍。
Ceralasertib (AZD-6738) 是由阿斯利康公司开发的,对ATR激酶的IC50值为1 nM, 目前已经进入临床II期研究, 用于治疗各种癌症(图3)。
M4344 (VX-803,Gartisertib) 最初由福泰制药(Vertex pharmaceuticals) 研究开发,代号为VX-803, 后被德国默克公司收购, 给予另外一个代号M4344(图3)。Gartisertib是一种ATP竞争性的,具有口服活性的,选择性的ATR抑制剂,Ki < 150 pM。
M1774是由德国默克子公司EMD Serono研究所研发的一种ATR激酶抑制剂。目前, 它作为单药或和PARP 抑制剂尼拉帕立治疗转移性或局部晚期不可切除实体瘤正在I 期临床试验中。截止到目前, M1774的结构式尚未公布。
ATRN-119是由美国宾夕法尼亚州的Atrin Pharmaceuticals开发的。该公司于2016年在美国癌症研究协会(American Association for Cancer Research, AACR)第107届年会上首次公布了ATRN-119的实验数据,用于治疗包括晚期实体瘤在内的癌症。
RP-3500是由加拿大的Repare Therapeutics开发的一种口服抑制剂。2020年7月,在晚期实体瘤患者中联合Talazoparib的I/IIa期试验对首批患者给药;2021年7月,在晚期实体瘤患者中开始了药物联合Niraparib或Olaparib的Ib/II期试验。
ART-0380是由Artios Pharma公司从美国MD Anderson癌症中心和中国Shang PharmaInnovation获得许可开发的一种新的口服ATR抑制剂。
拜耳公司开发的ATR抑制剂BAY1895344(Elimusertib)进行临床I期研究,用于实体瘤和淋巴瘤的研究(图3)。
2023年1月11日,北京泰德制药股份有限公司1类新药“TCC1727片”的临床试验申请获CDE受理。据公开资料,TCC1727是选择性ATR激酶抑制剂,靶向作用于DNA损伤修复(DDR)通路重要激酶ATR 。
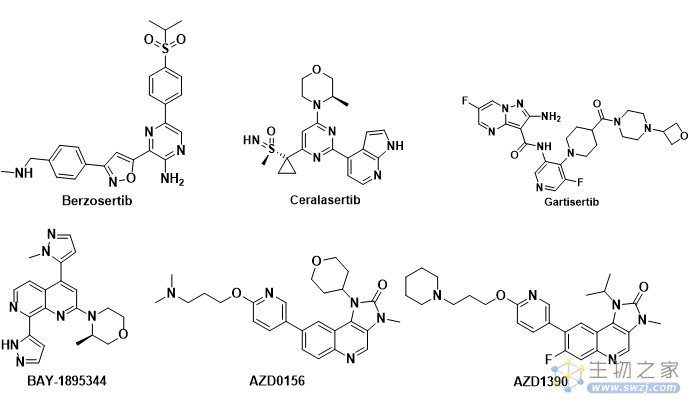
图3. ATR和ATM临床在研抑制剂
ATM进入临床研究的抑制剂有AZD0156和AZD1390(图3)。AZD0156/AZD1390是由阿斯利康开发的两款ATM抑制剂,两个分子的结构非常相似。AZD0156/AZD1390先后进入临床试验研究,随后AZD0156被终止临床;AZD1390目前正在开展临床试验研究。
AZD1390是一种具有口服活性的、能穿透中枢神经系统的ATM抑制剂,在细胞中IC50为0.78 nM。它对ATM的选择性是对PIKK家族其他相关酶的10,000以上,具有良好的选择性。
目前对AZD1390开展的临床研究有:(1)一项 0/1b 期、单中心临床试验,AZD1390 扩展期加分割放疗用于复发性 WHO IV 级胶质瘤患者(NCT05182905);(2)一项 I 期多中心研究,旨在评估 AZD1390 递增剂量联合放疗治疗多形性胶质母细胞瘤和实体瘤脑转移患者的安全性、耐受性和药代动力学(NCT03423628)。
DNA-PK抑制剂
DNA-PK对DSBs有反应,并在NHEJ修复途径中起关键作用(图4)。从机制上讲,Ku复合体(Ku70 和 Ku80)首先启动NHEJ。随后,DNA-PKcs(称为DNA依赖性蛋白激酶催化亚基)与DSB结合并启动DNA修复。其他NHEJ核心成分(例如DNA连接酶IV,Artemis,XRCC4,PAXX和XLF)以紧密对齐和连接两端,完成DNA修复过程。
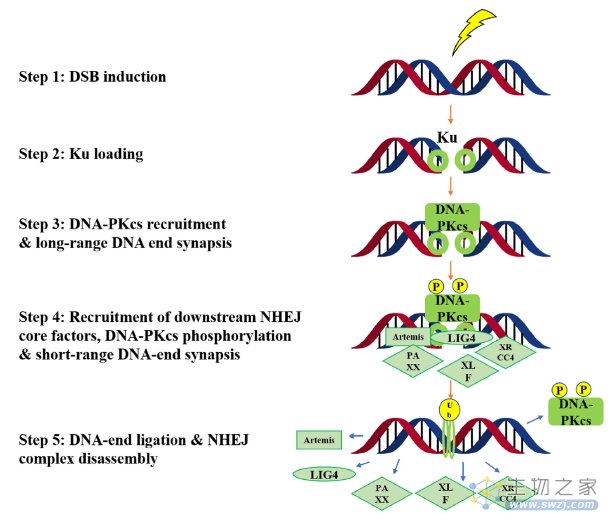
图4. DNA-PK在非同源末端连接修复途径中的生物学功能
DNA-PK抑制剂的开发主要集中在DNA-PKcs的催化活性上,而新的抗DNA-PKcs方法,如DNA-PKcs抑制性microRNA或靶向Ku异二聚体的抑制剂,则基于ATP结合位点的同源模型。目前,大多数DNA-PK抑制剂的临床研究(表1)集中在与癌症化疗或放疗联合使用的效果。
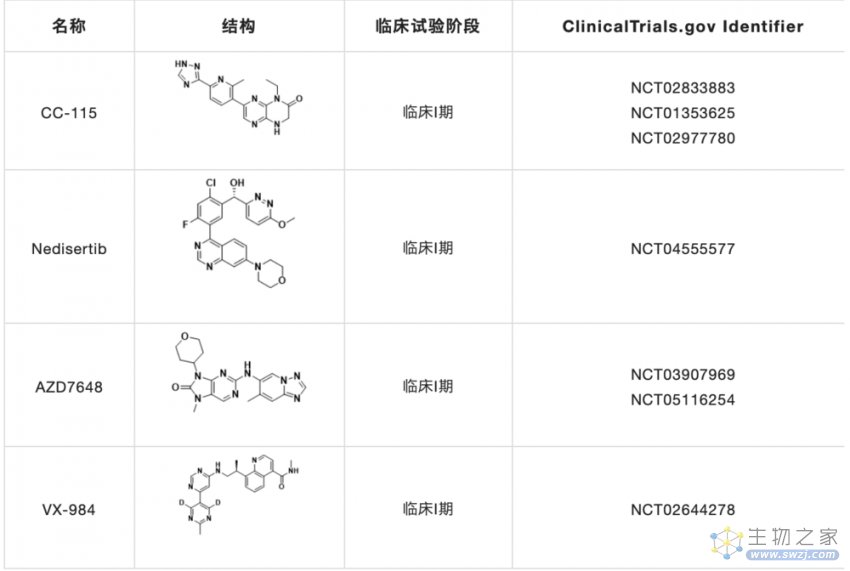
表1. 正在进行临床试验的DNA-PK抑制剂
CHK1和WEE1抑制剂
CHK1和WEE1在多种癌症中过表达(例如 HG-SOC高级别浆液性卵巢癌和乳腺癌)。当WEE1和/或CHK1被抑制时,肿瘤细胞会带着未修复的DNA过早进入有丝分裂,导致肿瘤细胞凋亡/死亡。
如图5所示,ss DNA可以首先激活ATR。然后活化的ATR可以磷酸化和激活CHK1,然后CDC25C和WEE1磷酸化,导致WEE1的激活和CDC25C的磷酸酶的失活。最后,WEE1可以磷酸化并灭活CDK1-cyclin B的复合物,从而导致G2相位停滞并为DNA修复提供时间
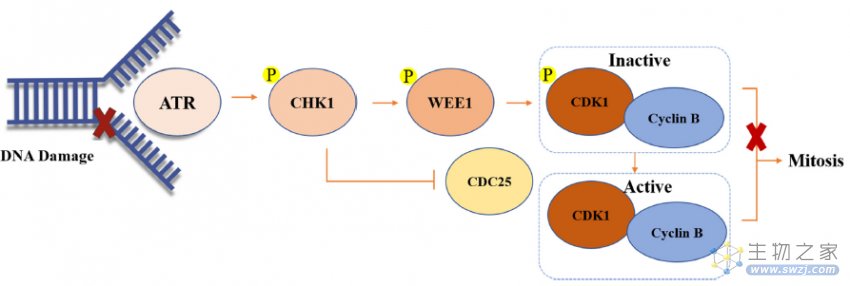
图5. CHK1和WEE1作用机制
目前CHK1研究在研的抑制剂主要有Prexasertib,GDC-0575和CCT245737(表2)。
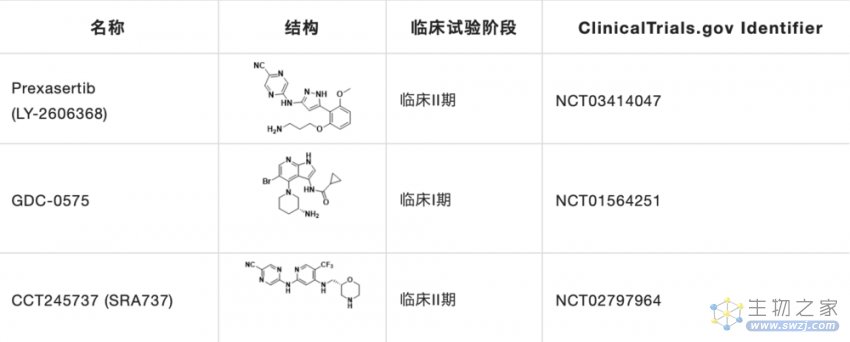
表2. 临床在研的小分子CHK1抑制剂
据不完全统计,目前在研的WEE1药物十余种,其中包括WEE1激酶抑制剂和PROTAC,表3是目前进入临床的WEE1抑制剂,研究进展最快的是进入临床II期的Adavosertib。其中WEE1激酶抑制剂按结构类型可分为嘧啶并吡啶酮类、嘧啶并吡唑酮类、咔唑并吡咯二酮类及其他类。
在研究企业上,其中AI制药公司Schrödinger,以及国外企业阿斯利康、辉瑞、Debiopharm等均在该领域布局;中国公司英派药业、首药控股、迪诺医药、智康弘仁也均在该领域进行布局。
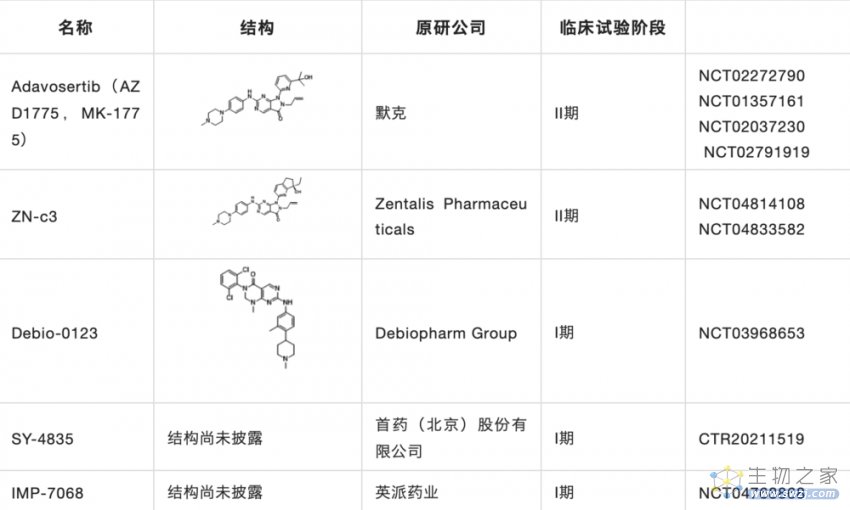
表3. 临床在研的小分子WEE1抑制剂
Marine等人报道了WEE1的PROTACs,基于WEE1抑制剂AZD1775和E3连接酶VHL配体或CRBN配体设计而得到的,如MA055和MA071,它们很明显的降解Wee1蛋白(图6)[3]。
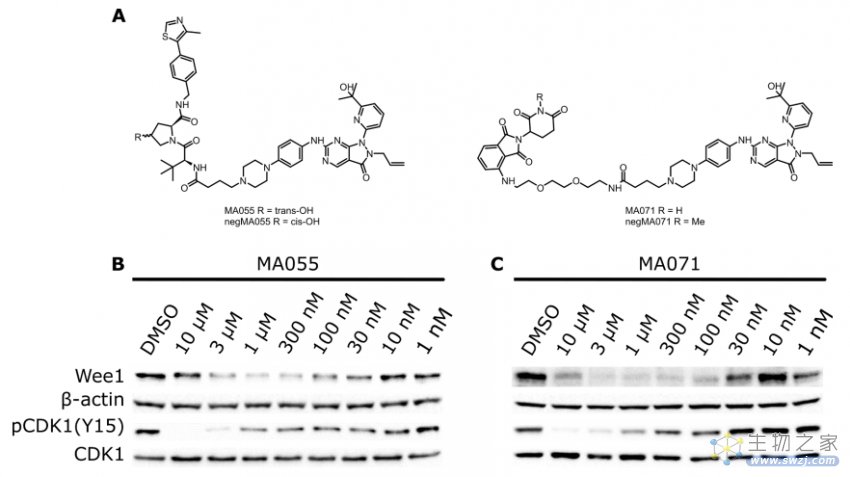
图6. WEE1的PROTACs
POLQ抑制剂
POLQ抑制剂目前研究较少,只有Artios Pharma Limited公司的ART4215进入临床二期研究(NCT04991480)。Artios Pharma Limited是一家领先的DNA损伤反应(DDR)公司,利用合成致死性开发广泛的精准药物产品线,用于治疗癌症。公司的研发管线有ATR抑制剂ART0380,POLQ抑制剂ART4215和RLT增敏剂等。
作为一种DNA聚合酶,POLQ参与修复DNA双链断裂。它在许多肿瘤中过表达,并在健康组织中低水平表达。研究表明,ART4215作为单药,在PARP抑制剂治疗后进展的患者中具有抗癌活性,它还可以与PARP抑制剂联用治疗未接受过PARP抑制剂治疗的患者,或与其它DNA损伤疗法(如电离辐射和细胞毒性化疗)联用。
Artios Pharma Limited公司开发的POLQ抑制剂主要围绕图6中的骨架结构来修饰优化,如改变A和D取代基结构,B环状结构,C用酰胺或者五环芳香环联系等(图7)。
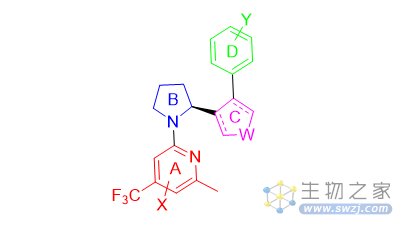
图7. Artios Pharma Limited公司开发的POLQ抑制剂结构类型
他们不仅申请了三篇专利WO2021028643A1,WO2021123785A1和WO2021028670A1,也在JMC详细报道了ART812的详细发现历程并且在nature communications上详细介绍了ART558和ART812的生物学功能,发现它们不仅诱导BRCA基因的合成致死,而且可以靶向由53BP1/Shieldin缺陷引起的PARPi抗性细胞。
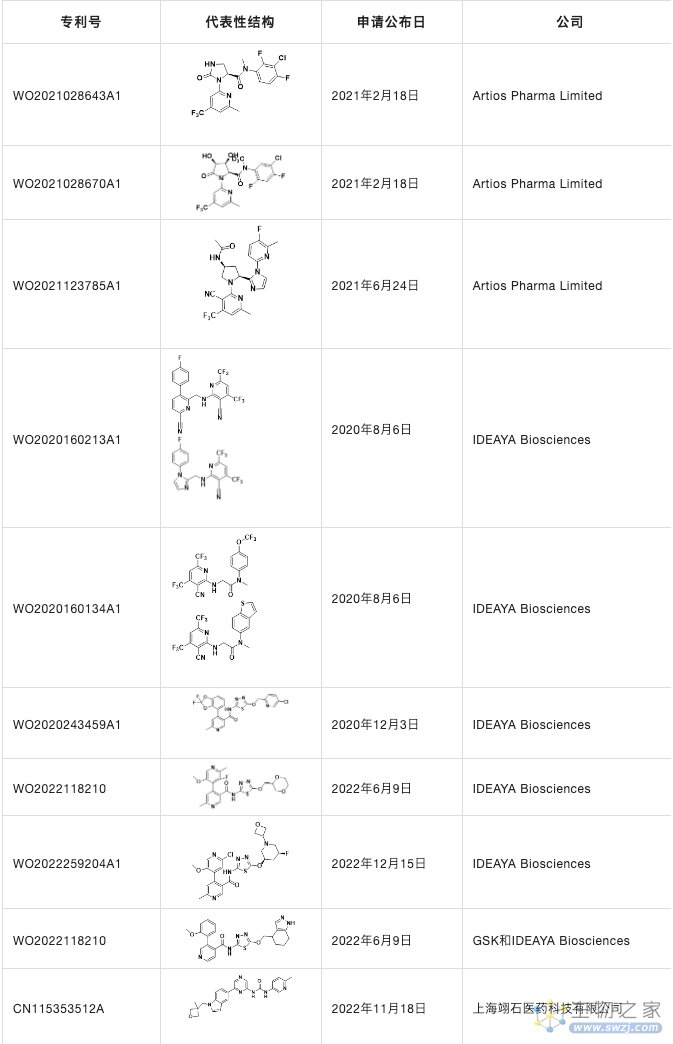
表4. POLQ抑制剂
除了Artios Pharma Limited公司之外,IDEAYA Biosciences也对POLQ抑制剂进行开发,主要围绕噻二唑骨架类结构,除此之外,国内的上海翊石医药科技有限公司也对POLQ靶点有所布局。
近期,Monica等人通过高通量筛选了350,000个化合物,发现苗头化合物,基于结构优化最终发现化合物RP-6685(图8),一种有效的、选择性的、口服生物可利用的POLQ抑制剂,在HCT116 BRCA2−/−小鼠肿瘤异种移植模型中显示疗效[4]。
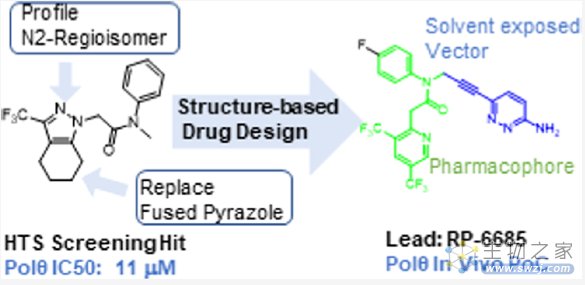
图8. 通过高通量筛选和基于结构优化发现化合物RP-6685
PARP抑制剂(PARPi)的耐药性的主要原因是BRCA1/2突变癌细胞HR修复活性的恢复。近期,研究报道SHLD1/2/3复合物的失活以及DSB末端切除术中其他蛋白(包括RIF1和REV7)的表达降低,恢复了HR的功能活性,降低了PARPi的疗效(图9)[5]。
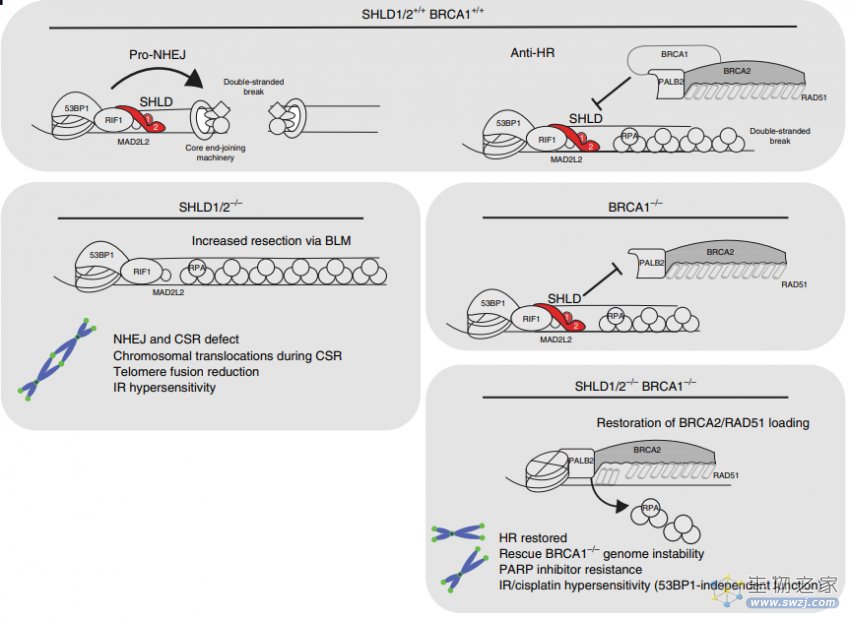
图9. SHLD1/2的敲除在BRCA1敲除细胞中导致对PARPi的耐药机制
针对这些对PARP抑制剂耐药的突变细胞株,研究发现POLQ抑制剂对这些细胞株敏感。Artios Pharma Limited公司在2021年2月18日披露一篇专利(WO2021028644A1),研究POLQ抑制剂在不同敲除细胞中的抑制作用,如REV7 KO SUM149,SHLD2 KO MDA-MB-436和SHLD2 KO HCC1395等。
小结
DNA损伤反应是引起科学界高度关注的最重要平台之一。基于DDR的癌症疗法能够通过合成致死相互作用为缺乏特定DDR功能的癌症患者量身定制治疗方案,从而提供潜在的更广泛的治疗窗口。基于DDR的癌症疗法最初与化疗联合使用,并且与不良不良反应有关,例如白细胞减少,胃肠道和骨髓毒性。
下一代DDR抑制剂的开发将侧重于通过改进选择性和特异性。开发DDR抑制剂的另一个挑战是探索更宽的治疗窗口和鉴定对DDR抑制敏感的遗传生物标志物。因此,开发DDR抑制剂领域是一个机遇与挑战共存的领域。
参考文献:
1.Binbin Cheng, Wei Pan, Yi Xing, Yao Xiao, Jianjun Chen, Zheng Xu, Recent advances in DDR (DNA damage response) inhibitors for cancer therapy, European Journal of Medicinal Chemistry 230 (2022) 114109
2.S. Matsuoka, B.A. Ballif, A. Smogorzewska, E.R. McDonald 3rd, K.E. Hurov, J. Luo, C.E. Bakalarski, Z. Zhao, N. Solimini, Y. Lerenthal, Y. Shiloh, S.P. Gygi, S.J. Elledge, ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage, Science 316 (2007) 1160e1166.
3.Marine C. Aublette, Tom A. Harrison, Elizabeth J. Thorpe, Morgan S. Gadd, Selective Wee1 degradation by PROTAC degraders recruiting VHL and CRBN E3 ubiquitin ligases, Bioorg. Med. Chem. Lett. 64 (2022) 128636.
4.Monica Bubenik, Pavel Mader, Philippe Mochirian, Frederic Vallee, Jillian Clark, Jean-François Truchon, Alexander L. Perryman, Victor Pau, Igor Kurinov, Karl E. Zahn, Marie-Eve Leclaire, Robert Papp, Marie-Claude Mathieu, Martine Hamel, Nicole M. Duffy, Claude Godbout, Matias Casas-Selves, Jean-Pierre Falgueyret, Prasamit S. Baruah, Olivier Nicolas, Rino Stocco, Hugo Poirier, Giovanni Martino, Alexanne Bonneau Fortin, Anne Roulston, Amandine Chefson, Stephane Dorich, Miguel St-Onge, Purvish Patel, Charles Pellerin, Stephane Ciblat, Thomas Pinter, Francis Barabe, Majida El Bakkouri, Paranjay Parikh, Christian Gervais, Agnel Sfeir, Yael Mamane, Stephen J. Morris, W. Cameron Black, Frank Sicheri, and Michel Gallant,Identification of RP-6685, an Orally Bioavailable Compound that Inhibits the DNA Polymerase Activity of Polθ,J. Med. Chem. 2022, 65, 19, 13198–13215
5.Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965.