50%以上罕见病的遗传学病因尚不清楚。
大规模人群队列的标准化基因组测序和表型分型有助于发现遗传学病因,但这需要高效且强大的分析方法。研究者建立了一个小型数据库“Rareservoir”,该数据里面是77,539名参与者的基因型和表型数据集合【来自10万人基因组测序计划(100,000 Genomes Project)】。
为了支持Rareservoir的构建过程,研究者开发了一个名为“rsvr”的补充软件包。该软件包有人群数据库(例如gnomAD)中的MAF信息、致病性评分(例如CADD评分)、预测序列本体(Sequence Ontology, SO)。
据估算,全球受罕见病影响的有2.63亿–4.46亿人,但只有少数患者接受了基因检测与诊断。总的来说,每20个人中就有1人罹患罕见病。目前,大约有1万种罕见病,但不到50%的罕见病找到了遗传学病因。对大量表型多样化的罕见病患者进行标准化基因组测序(GS),可以在广泛的数据中发现遗传学病因,同时提高患者的遗传学诊断率。10万人基因组计划(100KGP)是迄今为止对罕见病患者进行的最大GS研究。Rareservoir库中的77,539名参与者包括了34,523名罕见病患者(英国国家卫生服务)和43,016名未患病的家属。100KGP的相关基因型和表型数据,可以通过“Genomics England Research Environment”网站提供给研究人员。
研究者使用贝叶斯遗传关联方法(Bayesian genetic association),即BeviMed,来推断基因与269种罕见病之间的相关性。该研究确定了241个已知和19个之前未知的基因型与表型的相关性。
研究者通过搜索数据库中疾病相关的家系,并使用生物信息学和体外功能实验来验证并确定了三种新基因ERG, PMEPA1和GPR156与三种新的罕见病的相关性。
ERG基因相关的原发性淋巴水肿
ETS家族转录因子编码基因ERG的功能丧失变异(loss-of-function, LOF)导致显性遗传性原发性淋巴水肿(primary lymphoedema)。
原发性淋巴水肿是一种罕见的疾病,其特征是淋巴管发育异常或功能障碍,导至淋巴液在组织中堆积,引起水肿和其他相关症状。ERG基因编码血管内皮细胞基因表达的关键转录调节因子,这种转录因子在正常情况下对淋巴管发育和功能起着重要的调控作用。如果ERG发生功能丧失突变,则可能会影响淋巴管的正常发育和功能,导至淋巴液不能正常流动,从而引起原发性淋巴水肿。
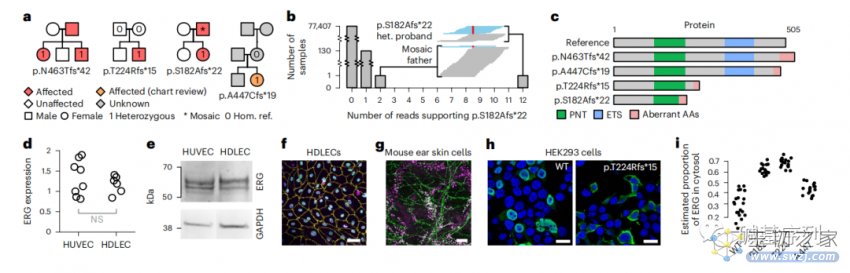
图a:携带ERG基因功能缺失变异的四名先证者先证物
图b:柱状图显示了100KGP中支持p.S182Afs*22 变异的reads数的分布。嵌入的窗口显示了该家系中两个携带p.S182Afs*22 (het)变异的患病成员在该位置的read序列堆积(即杂合型)。参考等位基因的reads是蓝色的,变异等位基因的reads是红色的。
图c:示意图显示了在cDNA和氨基酸水平上每个变异体的影响,以及与经典转录本相关的蛋白质产物。PNT:pointed domain;ETS:红细胞转化特异性DNA结合域;AA:氨基酸
图d:逆转录PCR检测人真皮淋巴内皮细胞(human dermal lymphatic endothelial cell, HDLEC)和人脐静脉内皮细胞 (human umbilical vein endothelial cell, HUVEC)中的ERG基因mRNA表达,两者无显著性差异。
图e:HUVEC和HDLEC中的蛋白裂解物的免疫印迹显示,ERG亚型对应的条带有相似强度表达。
图f:对培养的HDLECs的免疫荧光显微镜显示,ERG(绿色)与淋巴内皮细胞核标记PROX1(紫色)和细胞核标记DAPI(蓝色)共定位。这一发现在出生后3周的小鼠耳皮肤的免疫荧光染色所证实(图g)。
图h,i:HEK293细胞过表达野生型ERG和p.T224Rfs*15变异ERG的免疫荧光显微镜图像。在不表达内源性ERG的HEK293细胞中,过表达野生型ERG cDNA重现了在HDLEC和小鼠耳皮肤模型中。然而,ERG基因变异cDNA的过度表达导致ERG错误定位于细胞核外的细胞质中,阻止其与DNA结合,抑制其发挥作为转录因子的功能。
这些功能性试验证实了ERG在淋巴管内皮细胞核内的高水平表达,与淋巴管生成期间的转录调节功能一致。在原发性淋巴水肿患者中,无义介导的降解或错误定位导致的单倍剂量不足,引起的核内ERG可用性降低,最终导致淋巴管生成缺陷。
PMEPA1基因相关的Loeys-Dietz综合征
转化生长因子-β调节因子(transforming growth factor-β regulator)PMEPA1基因的最后一个外显子的截短变异导致Loeys-Dietz综合征。
Loeys-Dietz综合征是一种罕见的遗传性疾病,其特征包括动脉瘤、动脉破裂、软骨发育异常等,这些症状可能会对患者的生命造成威胁。PMEPA1的致病性变异与显性遗传性家族性胸主动脉瘤(Familial thoracic aortic aneurysm disease, FTAAD有)关。
在100KGP队列中,在三个欧洲裔的FTAAD家系中,发现同一个PMEPA1基因 p.S209Qfs*3变异。在八名日本裔FTAAD患者中,也发现了相同的变异。在100KGP Pilot Programme的另一个病例集中,也发现p.S209Afs*61变异;在比利时的一个FTAAD家系中,也发现p.P207Qfs*3。这些结果表明,PMEPA1基因最后一个外显子的截短突变会导致Loeys-Dietz综合征。
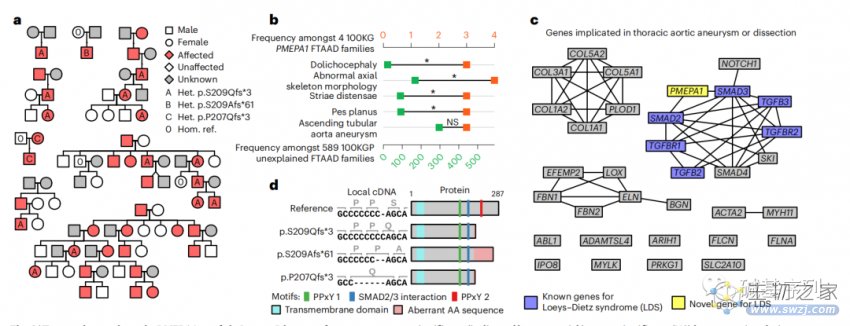
为了了解这个基因变异的分子机制,发现PMEPA1的蛋白-蛋白相互作用和“胸主动脉瘤或夹层”Panel App面板中完整的高可信基因。PMEPA1编码转化生长因子-β信号转导的负调节因子,而这一通路涉及多种主动脉疾病,包括Loeys-Dietz综合征。已知Loeys-Dietz综合征的基因编码TGF-β通路中紧密相互作用的蛋白亚群的一部分,其中SMAD2, SMAD3和PMEPA1编码的蛋白之间存在直接相互作用(图c)。截断变异发生在转录本的最后一个外显子,它们可能会逃避无义介导的降解。然而,他们截断作用会去掉PPxY相互作用的基序,而保持SMAD相互作用基序完整(图d),这可能影响PMEPA1和SMAD2/3之间的结合,并通过功能获得机制(gain-of-function)改变TGF-β信号传导。
GPR156基因相关的先天性听力障碍
GPR156基因的LOF导致隐性遗传性先天性听力障碍。
隐性遗传性先天性听力障碍是一种常见的遗传性疾病,其特征是患者从出生时就存在听力障碍,这可能会影响语言和认知的发展。这种听力障碍通常是由遗传因素引起的,可以是由单个基因突变引起的单基因遗传,也可以是由多个基因突变和环境因素共同作用引起的复杂遗传。
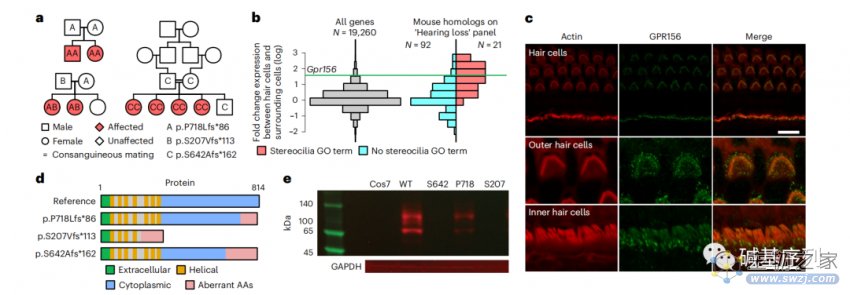
图a:在数据库中,研究在三个家系发现GPR156基因的3个变异位点(p.S207Vfs*113、p.P718Lfs*86、p.S642Afs*162)。在这三个家系中的八名患者均表现为先天性的非综合征性双侧感音神经性听力损失。
图b:静纤毛破坏是许多人类孟德尔耳聋疾病的共同致病机制,与周围细胞相比,毛细胞中GPR156的过表达与目前21个与静纤毛相关基因的过表达是相符。
图c:野生型小鼠的Corti和前庭系统的免疫荧光染色,发现GPR156与肌动蛋白强共定位于Corti器的外毛细胞和内毛细胞的顶端表面。
图d:p.S207Vfs*113变异位于GPR156基因的10个外显子,通过无义介导的GPR156基因的mRNA衰减,使得GPR156表达缺失。相比而言,p.S642Afs*162和p.P718Lfs*86变异均发生在GPR156最后一个外显子内,可能导致异常的GPR156表达(氨基酸序列改变和截短蛋白)。
图e:为了确定这些变异对蛋白表达的影响,用野生型GPR156的cDNA和三种突变型GPR156,转染Cos7细胞(不表达内源性GPR156),并标记绿色荧光蛋白(GFP)报告基因。野生型细胞能够表达GPR156-GFP融合蛋白,而突变型细胞没有明显表达该蛋白,或表达明显减少,这表明这三种截短蛋白都被降解了。
GPR156编码一种膜蛋白受体,这种受体在耳蜗和听神经中表达。GPR156中的双等位基因截断变异阻止GPR156蛋白的表达,破坏听觉上皮细胞中的静纤毛形成,影响耳蜗和听神经的正常发育和功能,从而引起先天性听力损失。
艾卡恩医学院西奈山分校遗传和基因组科学副教授Ernest Turro博士说:“虽然罕见疾病的发生率很低,但总体而言,它们非常普遍。找到剩余的病因对于我们理解人类生物学以及开发诊断和治疗方法非常重要。许多患有罕见疾病的人们在多年的努力下才能获得遗传学诊断。通过开发和应用统计方法和计算方法,寻找罕见疾病的新病因,我们希望扩大对这些疾病潜在病因的认识,缩短患者的诊断时间,并为治疗的开发铺平道路。”
研究的局限性
大约82%的100KGP参与者是欧洲裔,限制了识别其他人群特有的致病变异的能力。
在分析的260个病例集中,有28个是少于5个先证者,限制了识别相应疾病类型的能力,这说明继续招募极罕见病患者的必要性。
研究只考虑了编码基因中的SNV和插入缺失。非编码基因和基因组调控元件的结构变异和罕见变异的探索需要进一步研究。
研究将注意力集中在罕见病的单基因模型上,而某些罕见病的遗传学病因可能是多基因的。
单基因病临床表型的多样化可以用多基因效应来解释。
基于大规模人群数据库
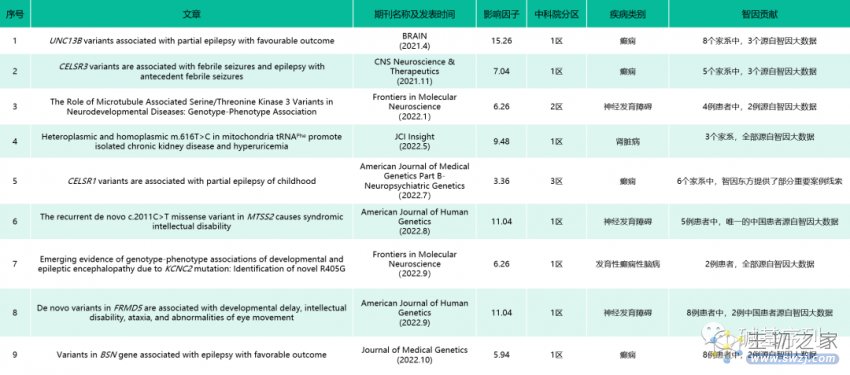
发现新基因的国内相关报道
通过基因型-表型的大规模人群数据库,发现新基因的研究,国内也有相关报道。
神经发育障碍新致病基因发现
智因东方拥有数十万中国人WES/WGS基因-表型数据集合,构建了智因大数据。2021年以来,智因大数据已经助力了6个新致病基因发现。
他们的10万级别的人群大数据助力了2个神经发育障碍的新致病基因发现,相关研究成果均发表于遗传学领域顶级期刊《美国人类遗传学杂志》。
由美国贝勒医学院、湖南省妇幼保健院牵头。研究报道了8例神经发育障碍患者,主要表现为发育迟缓、智力残疾、共济失调、癫痫发作和眼球运动异常等症状。8例患者的FRMD5基因均存在de novo错义变异,其中2个中国患者源自智因大数据。
由美国贝勒医学院牵头,联合中国人民解放军白求恩国际和平医院在内的多个研究机构共同发表。研究报道了5个表现为发育迟缓、智力障碍、眼动缺陷或失明、(相对)小头畸形,并有轻度面部畸形的患者。5个患者均存在相同的MTSS2变异p.Arg671Trp,其中唯一的中国患者源自智因大数据。
神经发育障碍新致病基因MAST4
湖南省儿童医院、湖南省妇幼保健院、卡尔加里大学多中心合作研究,首次报道了MAST4基因是神经发育障碍伴发育迟缓和婴儿痉挛的新致病基因。研究对一千多例神经发育障碍患儿进行了家系全外显子组测序,发现4例无亲缘关系、临床表现为神经发育障碍伴发育迟缓和婴儿痉挛症状、均携带MAST4基因de novo错义变异的患者(其中2例源自智因大数据)。
先天性小头畸形新致病基因
湖南省儿童医院、湖南省妇幼保健院科研团队共同开始发现了1例罕见的UGDH变异导致的发育性癫痫性脑病84型(DEE84,MIM:#618792,该疾病于2020年首次命名,发病率约1/1400万–1/200万),临床表现包括:发育迟缓、难治性癫痫、婴儿痉挛以及伴有面部畸形的张力减退。研究者在智因大数据的帮助下,又发现了另1例无亲缘关系的 UGDH 复合杂合变异致病患者。第2例患者除了有难治性癫痫、严重发育迟缓等症状外,同样表现出典型的先天性小头畸形。斑马鱼实验发现,ughd敲除斑马鱼表现为头长、体长和头/体长比例的显著降低,并且伴有中枢神经系统荧光面积的显著减少。这进一步证实了 UGDH/ughd 与小头畸形之间的基因-表型关联。
2022年,智因东方助力7个人类新致病基因的发现的重磅科研成果。
还有其他的
中国基因大数据计划和成果盘点
随着基因测序的不断发展,大规模人群数据库和生物信息分析能力是国内基因相关企业的最大竞争力。如何利用AI和大数据的研究进展,开发先进的生物信息算法和技术,去准确、高效地解读序列,识别和标识疾病相关的突变的同时,最大限度的实现流程自动化,减少人为误差,提高工作效率,对基因检测公司提出了非常高的要求。
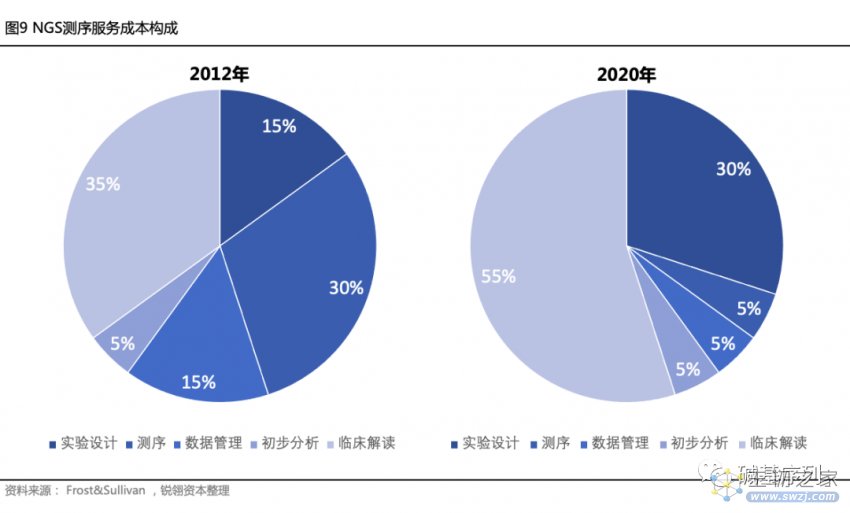
随着测序生产成本和生物信息基于云平台的分析成本不断降低,基因数据解读在基因检测服务中的成本比例将逐渐提高。根据Frost&Sullivan的NGS测序服务成本构成数据,从2013年到2020年,在超摩尔定律下测序仪在整个测序服务的成本占比下降了83.3%,而数据解读成本占比翻了一倍,占整体成本的30%。
来源:遗传病基因检测行业研究报告