由正常健康组织演变为良性肿瘤最后致恶性肿瘤的起始事件仍旧不得而知。目前,学界比较公认的是,上述演变是由基因突变所引起的【1】;然而,“在表型正常的上皮细胞中也存在可驱动细胞癌变的突变”却极大的挑战了上述观念【2】,因此在诱发细胞癌变方面考虑细胞和环境因素对癌变的产生和进展更为重要【3,4,5】。事实上,对于小鼠来说,无诱变(nonmutagenic)环境变化可以促使小鼠生成肿瘤【6,7】,而对于人类来说,持续的慢性炎症性环境也会增加罹患癌症的风险【8,9】。即使来自于同一组织的基因背景相同的细胞,上述现象也具有异质性【10】。遗传谱系示踪实验的结果类似,也就是这些细胞演变为肿瘤细胞和癌变的概率并不相同。这一异质性说明,在肿瘤生成过程中,突变的细胞要么具有,要么获得了可以改变细胞状态的能力,这一现象也被称之为细胞可塑性(cellular plasticity)。
发育、重塑以及病理可塑性的决定性因素在染色质层面,因为单个细胞的转录谱可以发生变化。具有高度可塑性的细胞,比如说干细胞,通常在发育过程中更加开放,或者说染色质可及性程度更高。之前有工作采用肺癌模型,通过对正常状态下的细胞进行去分化(de-differentiation),并进行单细胞基因组学检测,用来表征肿瘤细胞可塑性。当然,在肿瘤生成初期,尤其是在环境因素加快肿瘤生成的情况之下,这一细胞可塑性的形成机制仍旧不得而知。而研究细胞可塑性是如何被启动,原癌组织如何发生癌变,以及细胞可塑性如何参与早期肿瘤演化,可以极大助力肿瘤发育研究,尤其是肿瘤演化的最初阶段的认知。
近期,来自美国纽约Memorial Sloan Kettering Cancer Center的Scott W. Lowe和Dana Pe’er研究组在Science上发表题为Epigenetic plasticity cooperates with cell-cell interactions to direct pancreatic tumorigenesis的文章,就肿瘤发生的起始阶段进行了深入研究。

胰腺导管腺癌(Pancreatic ductal adenocarcinoma)确诊时往往为时已晚,已经缺乏治疗手段。目前认为,其发病是由遗传学和表观基因组重编程(epigenetic reprogramming events)共同决定的。与其它基因组高度异质性肿瘤类型不同的是,胰腺导管腺癌都是由原癌基因KRAS的活性突变所引起的。当然,KRAS突变的上皮细胞仍旧表型正常,并且,在炎性信号,比如胰腺炎(pancreatitis)刺激下,可以演化为肿瘤发生前(preneoplastic)或者是良性肿瘤病灶。
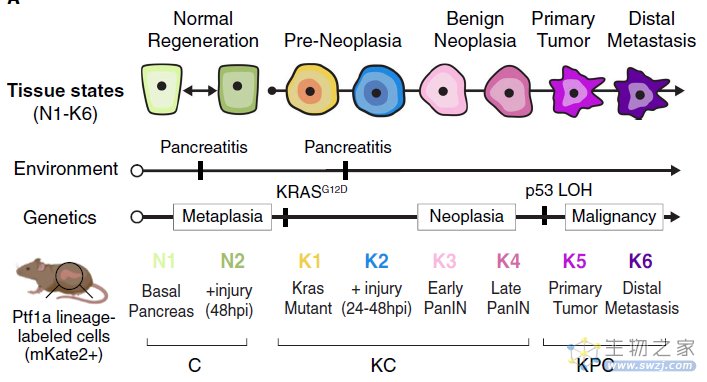
作者以及其它研究组之前的工作指出,原癌基因KRAS在没有进一步突变的情况之下,叠加炎症信号可以驱动大批量染色质重塑,从而促使肿瘤发生。当然,此处存在一个重要问题,这就是KRAS介导的细胞可塑性是如何参与健康细胞(良性组织)转变为恶性疾病的?这一过程之中的胞内和胞外决定细胞可塑性,以及最终演变为癌细胞状态的关键因素是什么?厘清上述问题的答案可以有效干预肿瘤发生。
为了研究胰腺导管腺癌的肿瘤可塑性(neoplastic plasticity),作者采用功能性修饰的基因改造小鼠模型(genetically engineered mouse models,简称GEMMs)模拟人类疾病的各个阶段,通过单细胞测序技术和计算生物学方法,比较了生理状态下、炎症期、癌变前期以及癌变细胞的异质性。作者再次在单细胞水平上确定了诱导肿瘤发生并与染色质重塑(chromatin remodeling)相关的异质化细胞状态。作者还通过干扰试验,揭示了数个信号模块和组织水平的相互作用网络,包括不同上皮和免疫细胞群之间的肿瘤驱动负反馈网络,并进一步通过小鼠验证其真实性。
综上所述,作者的工作展示了肿瘤特异性组织重塑(tissue-remodeling)过程,有助于加深对胰腺导管腺癌病理的理解。
文章来源
https://doi.org/10.1126/science.add5327
参考文献
1. B. Vogelstein et al., Cancer genome landscapes. Science 339, 1546–1558 (2013). doi: 10.1126/science.1235122; pmid: 23539594
2. I. Martincorena et al., High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). doi: 10.1126/science.aaa6806;
pmid: 25999502
3. N. Wijewardhane, L. Dressler, F. D. Ciccarelli, Normal somatic mutations in cancer transformation. Cancer Cell 39, 125–129 (2021). doi: 10.1016/j.ccell.2020.11.002; pmid: 33220180
4. D. Hanahan, Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022). doi: 10.1158/2159-8290.CD-21-1059; pmid: 35022204
5. A. S. Nam, R. Chaligne, D. A. Landau, Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 22, 3–18 (2021). doi: 10.1038/ s41576-020-0265-5; pmid: 32807900
6. C. Guerra et al., Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302 (2007). doi: 10.1016/ j.ccr.2007.01.012; pmid: 17349585
7. C. Carrière, A. L. Young, J. R. Gunn, D. S. Longnecker, M. Korc, Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem. Biophys. Res. Commun. 382, 561–565 (2009). doi: 10.1016/ j.bbrc.2009.03.068; pmid: 19292977
8. A. B. Lowenfels et al., Hereditary pancreatitis and the risk of pancreatic cancer. J. Natl. Cancer Inst. 89, 442–446 (1997). doi: 10.1093/jnci/89.6.442; pmid: 9091646
9. L. M. Coussens, Z. Werb, Inflammation and cancer. Nature 420, 860–867 (2002). doi: 10.1038/nature01322;
pmid: 12490959
10. V. Giroux, A. K. Rustgi, Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 17, 594–604 (2017). doi: 10.1038/nrc.2017.68; pmid: 28860646