传统上,人类疾病被视作单一、独立的实体来研究,这限制了研究人员将人类疾病视为一个复杂的、内稳态系统中的相互依赖状态。这项研究使用超过 1.51 亿美国人的时间戳临床记录,构建隐空间,将疾病表征为一个连续高维空间中的点,使得具有相似病因和表型的疾病相互靠近。

研究中使用 UK biobank 队列,其中包含 50 万参与者,执行一个针对新定义的反映个人健康状况的人类数量性状的全基因组关联分析,对应于疾病空间中的患者位置。研究发现了涉及 108 个基因位点的 116 个基因关联,然后使用嵌入空间中疾病聚类分析产生的 10 个疾病聚簇以及 30 种常见疾病,证明这些基因关联可用于稳健地预测各种疾病。
该研究的第一步,是从包含了 547 种疾病,涵盖 1.51 亿美国人的医疗数据中,使用 word2vec 词向量的方式,构建疾病的隐空间。初始数据包括描述医疗诊断及发病场景的文本。通过构建语言模型,预测诊断记录中的下一个词,构建的隐空间包含了疾病之间的相互关系。降维到 20 维后,可以看到不同类型的疾病之间的差异(图 1bc)。进一步降维到 3 维时,可以看到不同类型的疾病出现聚簇(图 1a)。
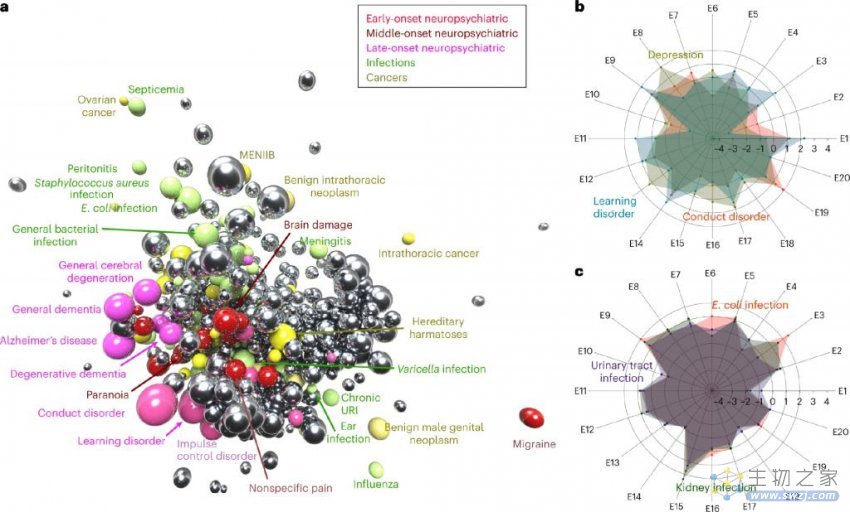
图 1. 基于疾病表型聚簇的可视化
基于医疗数据,研究者将疾病进行聚类,得到了 20 个聚簇。图中的不同颜色代表了人体的不同系统,图中标出文字的疾病,代表了该疾病与对应聚簇的 cos 相似性的大小,每个聚簇列出最相关的 10 种疾病。
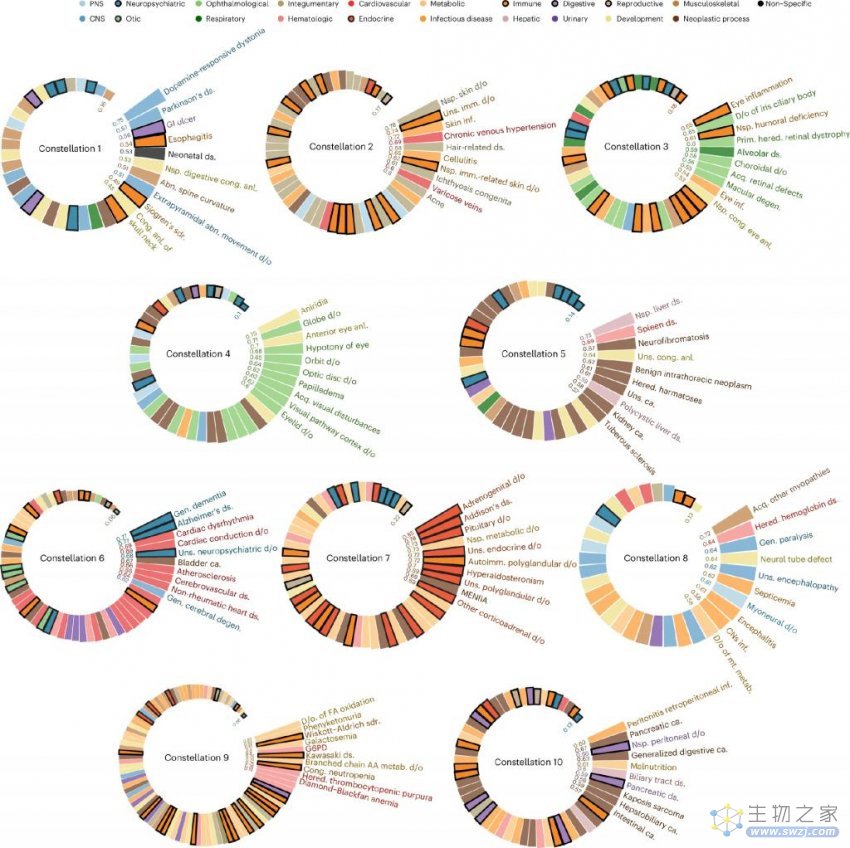
图 2. 疾病聚簇的结果图
在构建了表征多种复杂疾病的隐空间后,使用既有表型数据、又有基因数据的 UK biobank 数据,根据医疗记录在隐空间中的表征,进行全基因组关联分析,预估每个维度上的遗传度(图 3a)。图 3b 展示了全基因组关联分析中存在统计显著的维度(标星),其中的颜色代表不同维度间的相关性。图 3c 为存在显著差异的基因位点的曼哈顿图。
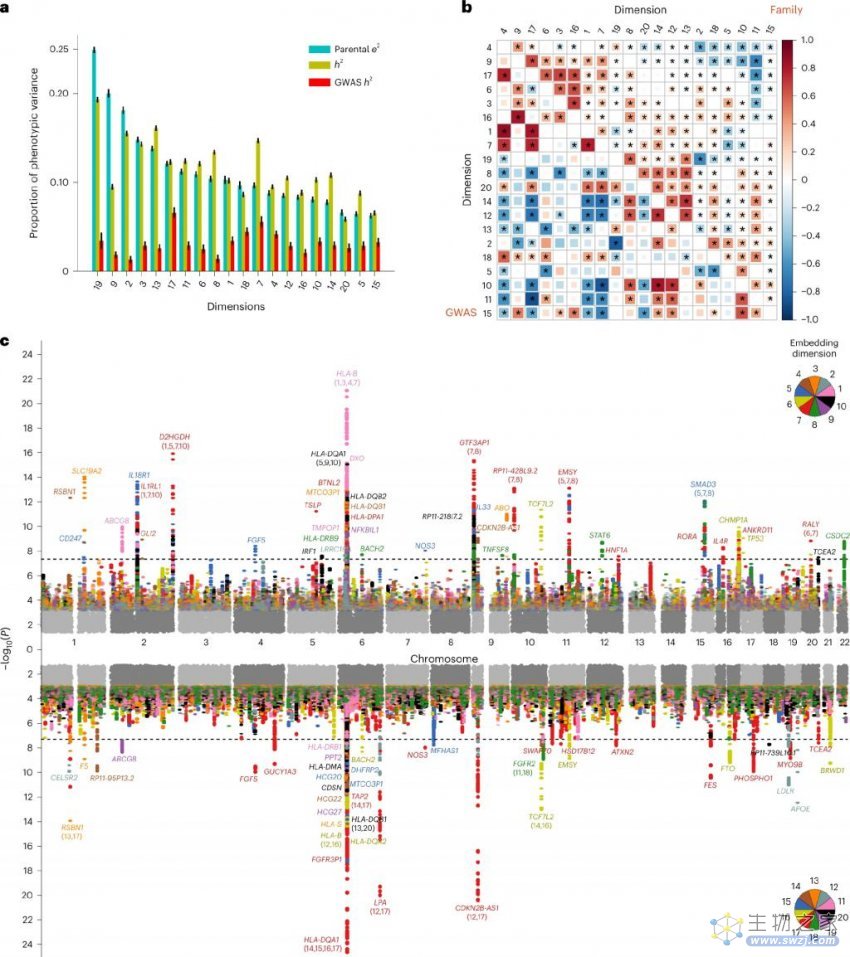
图 3. 对于疾病在隐空间中表现聚类进行全基因组关联分析的结果
在全基因组关联分析后,可基于找到的表型相关基因位点,进行多基因风险评估,结果显示,除了对于变应型鼻炎,基于新方法得到的预测比基于单一疾病的方法准确度更高。这说明将疾病视作一个整体研究,有助于把握住疾病间的相互关系。该研究还指出了自然语言处理的方法,可用于临床诊断数据,从中获得关于生物复杂性的洞见。